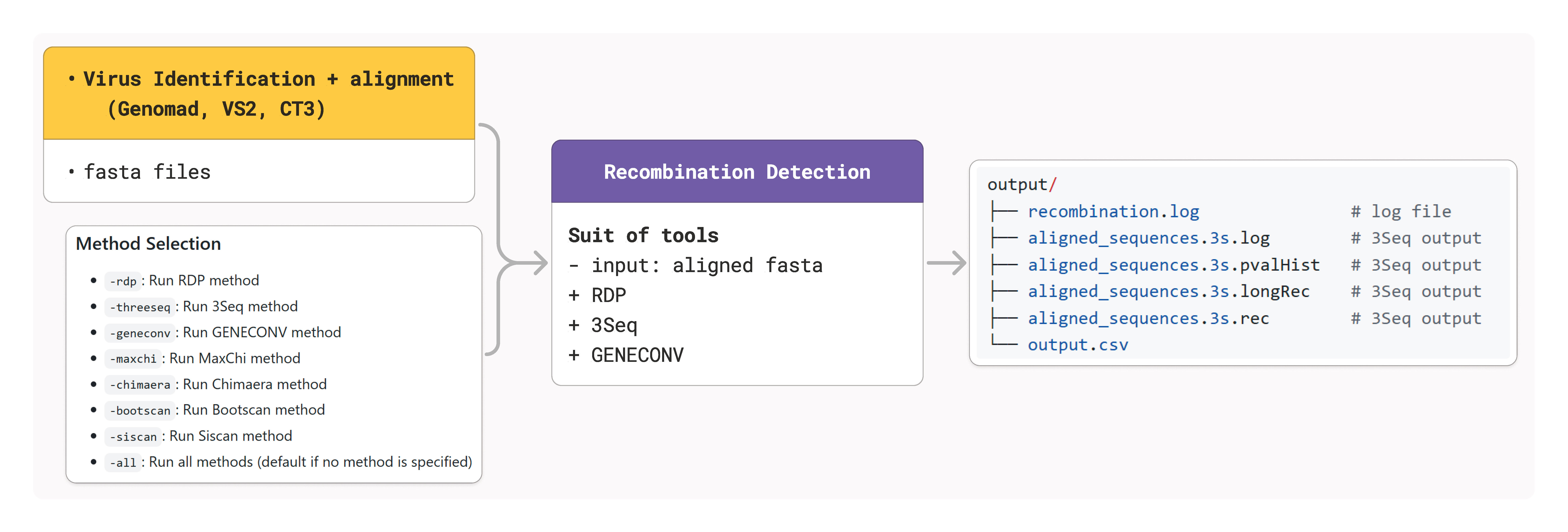
Recombination Detection Module
The recombination module detects recombination events in DNA sequences using multiple computational methods integrated through OpenRDP. It provides comprehensive recombination analysis using up to seven different detection algorithms.
Overview
Recombination is a crucial evolutionary process in viral genomes that can:
Generate genetic diversity
Create new viral strains
Influence host range evolution
Affect vaccine and therapeutic development
The recombination module implements multiple detection methods to ensure robust identification of recombination events.
Detection Methods
CRESSENT integrates seven recombination detection methods:
Primary Methods
RDP: Identifies recombination by detecting unusual phylogenetic relationships
GENECONV: Detects gene conversion events using statistical analysis
Bootscan: Uses bootstrap values to identify recombination breakpoints
MaxChi: Maximum chi-square method for breakpoint detection
Chimaera: Detects recombination using multiple reference sequences
3Seq: Three-sequence method for recombination detection
Siscan: Sister-scanning method for recombination identification
Method Reliability
Events detected by multiple methods (≥3) are considered highly reliable:
Algorithm Workflow
Sequence Preprocessing: Validates alignment quality and sequence integrity
Method Execution: Runs selected recombination detection algorithms
Statistical Analysis: Calculates p-values for detected events
Result Integration: Combines outputs from multiple methods
Significance Filtering: Applies statistical thresholds for reliable detection
Usage
Run All Methods
Detect recombination using all available methods:
cressent recombination \
-i aligned_sequences.fasta \
-o output/recombination \
-f recombination_results.csv \
--all
Run Specific Methods
Run selected methods for targeted analysis:
cressent recombination \
-i aligned_sequences.fasta \
-o output/recombination \
-f recombination_results.csv \
-rdp -bootscan -maxchi
Custom Configuration
Use custom parameters via configuration file:
cressent recombination \
-i aligned_sequences.fasta \
-o output/recombination \
-f recombination_results.csv \
-c custom_config.ini \
--all
Parameters
Required Parameters
-i, --input: Input alignment file in FASTA format-o, --output: Output directory for results-f, --output_file: Output CSV file name for results
Method Selection
-rdp: Run RDP method-threeseq: Run 3Seq method-geneconv: Run GENECONV method-maxchi: Run MaxChi method-chimaera: Run Chimaera method-bootscan: Run Bootscan method-siscan: Run Siscan method-all: Run all available methods
Optional Parameters
-c, --config: Configuration file for method parameters-quiet: Suppress console output-verbose: Enable detailed logging
Output Format
The recombination analysis produces a comprehensive CSV file with the following structure:
Column |
Description |
|---|---|
Method |
Detection method used |
Recombinant |
Sequence identified as recombinant |
Major_Parent |
Predicted major parent sequence |
Minor_Parent |
Predicted minor parent sequence |
Breakpoint_Start |
Start position of recombination region |
Breakpoint_End |
End position of recombination region |
Pvalue |
Statistical significance of detection |
Multiple_Comparisons |
Corrected p-value |
Example Output
Method |
Recombinant |
Major_Parent |
Minor_Parent |
Breakpoint_Start |
Breakpoint_End |
Pvalue |
Multiple_Comparisons |
|---|---|---|---|---|---|---|---|
RDP |
Sequence_A |
Sequence_B |
Sequence_C |
245 |
678 |
0.0023 |
0.0156 |
Bootscan |
Sequence_A |
Sequence_B |
Sequence_C |
240 |
685 |
0.0034 |
0.0204 |
MaxChi |
Sequence_A |
Sequence_B |
Sequence_C |
250 |
670 |
0.0019 |
0.0133 |
Result Interpretation
Statistical Significance
P-value < 0.05: Statistically significant recombination event
P-value < 0.01: Highly significant recombination event
Multiple methods: Events detected by ≥3 methods are most reliable
Breakpoint Analysis
Examine breakpoint positions to understand:
Recombination hotspots: Regions with frequent breakpoints
Functional domains: Impact on protein function
Phylogenetic implications: Effect on evolutionary relationships
Data Analysis Workflow
Python Analysis Example
import pandas as pd
import matplotlib.pyplot as plt
# Load recombination results
df = pd.read_csv("recombination_results.csv")
# Filter significant events
significant = df[df['Pvalue'] < 0.05]
# Count methods per recombinant
method_counts = significant.groupby('Recombinant')['Method'].value_counts().unstack(fill_value=0)
method_counts['Total_Methods'] = method_counts.sum(axis=1)
# Identify reliable events (≥3 methods)
reliable_events = method_counts[method_counts['Total_Methods'] >= 3]
print("Reliable recombination events:")
print(reliable_events)
# Plot breakpoint distribution
plt.figure(figsize=(10, 6))
plt.hist(significant['Breakpoint_Start'], bins=20, alpha=0.7)
plt.xlabel('Breakpoint Position')
plt.ylabel('Frequency')
plt.title('Distribution of Recombination Breakpoints')
plt.show()
Integration with Phylogenetic Analysis
Recombination detection should be performed before phylogenetic analysis:
# 1. Align sequences
cressent align \
--input_fasta sequences.fasta \
-o alignment/
# 2. Detect recombination
cressent recombination \
-i alignment/sequences_aligned_trimmed_sequences.fasta \
-o recombination/ \
-f recomb_results.csv \
--all
# 3. Analyze results and potentially remove recombinant sequences
# 4. Build phylogenetic trees with cleaned dataset
cressent build_tree \
-i cleaned_alignment.fasta \
-o phylogeny/
Best Practices
Input Preparation
High-quality alignment: Ensure proper sequence alignment before analysis
Sufficient diversity: Include adequate sequence diversity for detection
Appropriate length: Sequences should be long enough to detect meaningful events
Method Selection
All methods: Use all methods for comprehensive analysis
Cross-validation: Require detection by multiple methods for reliability
Statistical thresholds: Apply appropriate p-value cutoffs
Result Validation
Manual inspection: Examine alignments around detected breakpoints
Phylogenetic analysis: Compare trees before/after recombinant removal
Functional analysis: Consider impact on protein function
Common Applications
Outbreak Investigation
Trace recombination events in epidemic strains
Identify parent strains in recombinant viruses
Understand transmission dynamics
Vaccine Development
Identify stable genomic regions for vaccine targets
Assess recombination risk in vaccine strains
Monitor vaccine escape variants
Troubleshooting
Common Issues
No Events Detected Check alignment quality and sequence diversity. Ensure adequate evolutionary distance.
Binary Compilation Errors The module automatically compiles required binaries. Check system compatibility and dependencies.
Memory Issues Reduce dataset size or increase available memory. Some methods are computationally intensive.
Statistical Significance Adjust p-value thresholds based on study requirements and multiple testing considerations.
Performance Considerations
Dataset size: Larger datasets require more computational time
Sequence length: Longer sequences provide more power but increase runtime
Method selection: Running all methods increases accuracy but computational cost
Parallel processing: Some methods can utilize multiple CPU cores
Example Complete Analysis
#!/bin/bash
# Complete recombination analysis workflow
echo "Starting recombination analysis..."
# 1. Prepare alignment
cressent align \
--threads 24 \
--input_fasta viral_genomes.fasta \
-o analysis/alignment
# 2. Run comprehensive recombination detection
cressent recombination \
-i analysis/alignment/viral_genomes_aligned_trimmed_sequences.fasta \
-o analysis/recombination \
-f comprehensive_recombination.csv \
--all \
--verbose
# 3. Generate summary report
echo "Analysis complete. Results in analysis/recombination/"
echo "Check comprehensive_recombination.csv for detailed results"