Secondary Structure Detection
CRESSENT provides specialized tools for detecting and analyzing secondary structures in CRESS DNA viruses, focusing on two critical elements: stem-loop structures and iterons. These features are essential for understanding viral replication mechanisms and genomic organization.
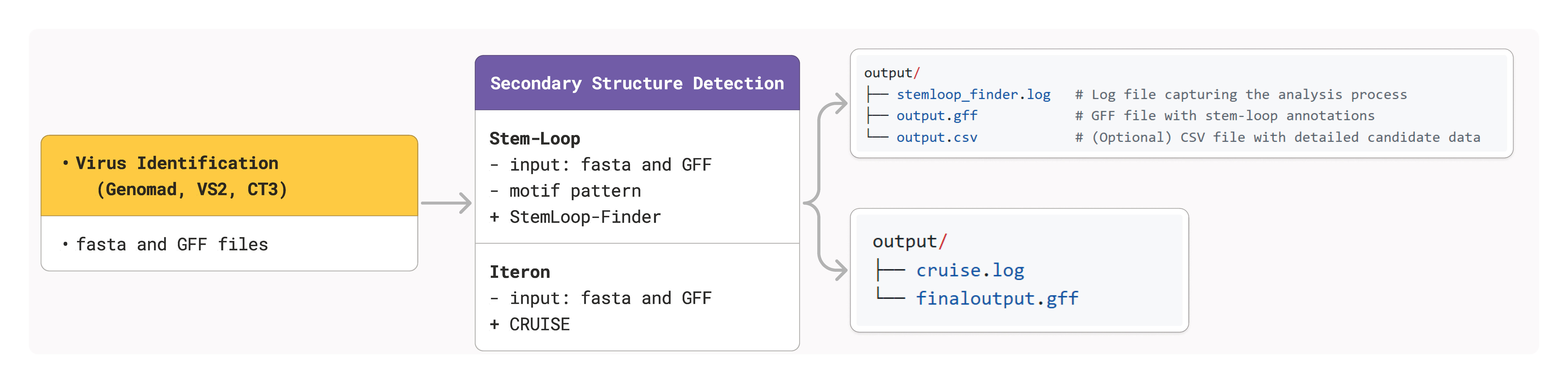
Overview
The secondary structure module includes:
Stem-loop detection using RNA folding algorithms and motif conservation
Iteron identification with CRUISE integration for replication origins
Structural validation through energy minimization and pattern recognition
Annotation output in standard GFF3 format
Stem-Loop Detection
Purpose
Identify hairpin structures with conserved motifs, particularly important for viral replication and packaging signals.
Basic Usage
cressent sl_finder -i sequences.fasta \
--gff_in annotations.gff \
--out_gff stemloops.gff \
--output sl_analysis/
Algorithm Overview
The stem-loop finder uses a multi-step approach:
Motif searching for conserved sequences (e.g., nonanucleotides)
RNA folding using ViennaRNA package
Structure parsing to identify stem-loop patterns
Validation based on stem/loop length criteria
Scoring using structural and sequence features
Parameters
Parameter |
Description |
Default |
|---|---|---|
|
Conserved motif pattern |
nantantan |
|
CRESS viral family |
None |
|
Ideal stem length |
11 |
|
Ideal loop length |
11 |
|
Bases around motif for folding |
15 |
|
Output CSV filename |
None |
Example Usage by Family
# Geminivirus analysis
cressent sl_finder -i gemini_genomes.fasta \
--gff_in annotations.gff \
--family geminiviridae \
--out_gff gemini_stemloops.gff \
--output gemini_analysis/
# Custom motif analysis
cressent sl_finder -i sequences.fasta \
--gff_in annotations.gff \
--motif "TAGTATTAC" \
--idealstemlen 12 \
--ideallooplen 8 \
--out_gff custom_stemloops.gff \
--output custom_analysis/
Output Files
GFF annotations (
stemloops.gff): Stem-loop and nonanucleotide featuresCSV results (
results.csv): Detailed structural informationLog file (
sl_finder.log): Processing details and statistics
GFF Output Format
##gff-version 3
sequence_01 sl_finder stem_loop 150 201 . + . Name=stem-loop
sequence_01 sl_finder nonanucleotide 165 173 . + . Name=nona
sequence_02 sl_finder stem_loop 89 145 . - . Name=stem-loop
sequence_02 sl_finder nonanucleotide 110 118 . - . Name=nona
CSV Output Format
seqID |
matched |
motif_start |
stem_start |
stem_end |
score |
folded_structure |
|---|---|---|---|---|---|---|
sequence_01 |
TAGTATTAC |
165 |
150 |
201 |
8.5 |
(((((…….)))))….. |
sequence_02 |
TAGTATTAC |
110 |
89 |
145 |
6.2 |
(((((……..)))))….. |
Iteron Detection
Purpose
Identify iterons (direct repeats) that serve as replication origins in CRESS DNA viruses, using the CRUISE algorithm.
Basic Usage
cressent run_cruise --input_fasta sequences.fasta \
--inputGFF annotations.gff \
--output cruise_analysis/ \
--outputGFF iterons.gff
CRUISE Algorithm
CRUISE (CRUcivirus Iteron SEarch) implements:
Substring enumeration within specified length ranges
Distance analysis between repeat occurrences
Stem-loop context validation
Scoring systems based on repeat quality and distribution
Known iteron database comparison
Parameters
Parameter |
Description |
Default |
|---|---|---|
|
Minimum iteron length |
5 |
|
Maximum iteron length |
12 |
|
Search range around nonanucleotide |
65 |
|
Use ranking system |
True |
|
Number of top iterons to report |
5 |
|
Maximum score for non-ranked mode |
40 |
|
Length-distance tolerance |
5 |
|
Optimal iteron length |
11 |
|
Maximum distance between iterons |
20 |
|
Optimal distance between iterons |
10 |
Known Iterons Database
CRUISE includes a database of characterized iterons:
# Examples of known iterons
TTGTCCAC # RDHV
AGTGGGA # Various circoviruses
GCCACCC # Begomovirus
GGGGA # Mastrevirus
TCTGA # Curtovirus
Advanced Parameters
cressent run_cruise --input_fasta sequences.fasta \
--inputGFF annotations.gff \
--output detailed_analysis/ \
--minLength 8 \
--maxLength 15 \
--range 100 \
--numberTopIterons 10 \
--maxDist 25 \
--bestDist 12 \
--scoreRange 30
Output Files
Annotated GFF (
iterons.gff): Original GFF with iteron annotationsProcessing log (
cruise.log): Detailed analysis logStatistics summary: Iteron discovery rates and patterns
Iteron Annotation Types
The CRUISE module annotates several types of features:
Feature Type |
Description |
|---|---|
|
Standard direct repeats |
|
Iterons associated with stem-loops |
|
Known/characterized iterons |
|
Database-matched sequences |
Combined Secondary Structure Analysis
Integrated Workflow
# Step 1: Identify stem-loops
cressent sl_finder -i cress_genomes.fasta \
--gff_in base_annotations.gff \
--family circoviridae \
--out_gff stemloops.gff \
--csv_out stemloop_details.csv \
--output structure_analysis/
# Step 2: Find iterons in context
cressent run_cruise --input_fasta cress_genomes.fasta \
--inputGFF structure_analysis/stemloops.gff \
--output structure_analysis/ \
--outputGFF complete_structures.gff \
--minLength 6 \
--maxLength 14 \
--numberTopIterons 8
Quality Control Pipeline
# Conservative stem-loop detection
cressent sl_finder -i sequences.fasta \
--gff_in annotations.gff \
--idealstemlen 10 \
--ideallooplen 9 \
--frame 20 \
--out_gff conservative_stemloops.gff \
--output qc_analysis/
# Comprehensive iteron search
cressent run_cruise --input_fasta sequences.fasta \
--inputGFF qc_analysis/conservative_stemloops.gff \
--output qc_analysis/ \
--minLength 4 \
--maxLength 16 \
--range 80 \
--numberTopIterons 15 \
--maxDist 30
Validation and Quality Assessment
Structural Validation
Energy scoring using ViennaRNA folding energies
Length criteria for biologically relevant structures
Motif conservation across related sequences
Distance constraints for iteron spacing
Quality Metrics
# High-confidence stem-loops
- Stem length: 8-15 bp
- Loop length: 6-20 bp
- Folding score: < 10
- Motif match: exact or 1 mismatch
# Validated iterons
- Length: 6-12 bp
- Occurrences: ≥ 2
- Distance: 8-15 bp
- Context: within 65 bp of nonanucleotide
Biological Interpretation
Stem-Loop Functions
Replication origins (often contain nonanucleotides)
Packaging signals for viral DNA
Regulatory elements for gene expression
Structural domains in viral genomes
Iteron Significance
Replication initiation sites
Rep protein binding domains
Copy number control elements
Evolutionary markers for virus classification
Best Practices
Input Preparation
Quality sequences with minimal gaps or ambiguities
Proper annotations in GFF3 format
Family-specific parameters when known
Consistent naming for sequence identifiers
Parameter Optimization
Family-appropriate motifs for stem-loop detection
Realistic length ranges for iteron searches
Contextual distances based on genome organization
Scoring thresholds appropriate for data quality
Validation Strategies
Cross-reference with known structures
Phylogenetic consistency across related sequences
Experimental validation when possible
Literature comparison for characterized viruses
Troubleshooting
Common Issues
No stem-loops detected:
Verify motif pattern accuracy
Check sequence quality and format
Adjust folding parameters
Consider alternative viral families
Excessive iteron predictions:
Increase scoring stringency
Reduce maximum distance parameters
Use ranking mode for top candidates
Filter by stem-loop context
ViennaRNA errors:
Check sequence format (DNA/RNA)
Verify installation completeness
Monitor memory usage for large sequences
Consider sequence length limitations
Performance Optimization
Large genomes:
Process in smaller segments
Increase memory allocation
Use parallel processing where available
Consider preliminary filtering
Parameter tuning:
Start with default values
Adjust based on known biology
Validate with characterized examples
Document parameter choices
Error Diagnostics
# Detailed logging output
INFO - Processing record: sequence_01
INFO - Found stem-loop at 150-201 in sequence_01
WARNING - No iterons found in sequence_02
ERROR - ViennaRNA folding failed: sequence too short