Visualization Overview
CRESSENT provides a comprehensive suite of visualization tools designed to create publication-ready figures for genomic and phylogenetic analysis. The visualization modules transform complex analytical results into clear, interpretable graphics suitable for scientific publications and presentations.
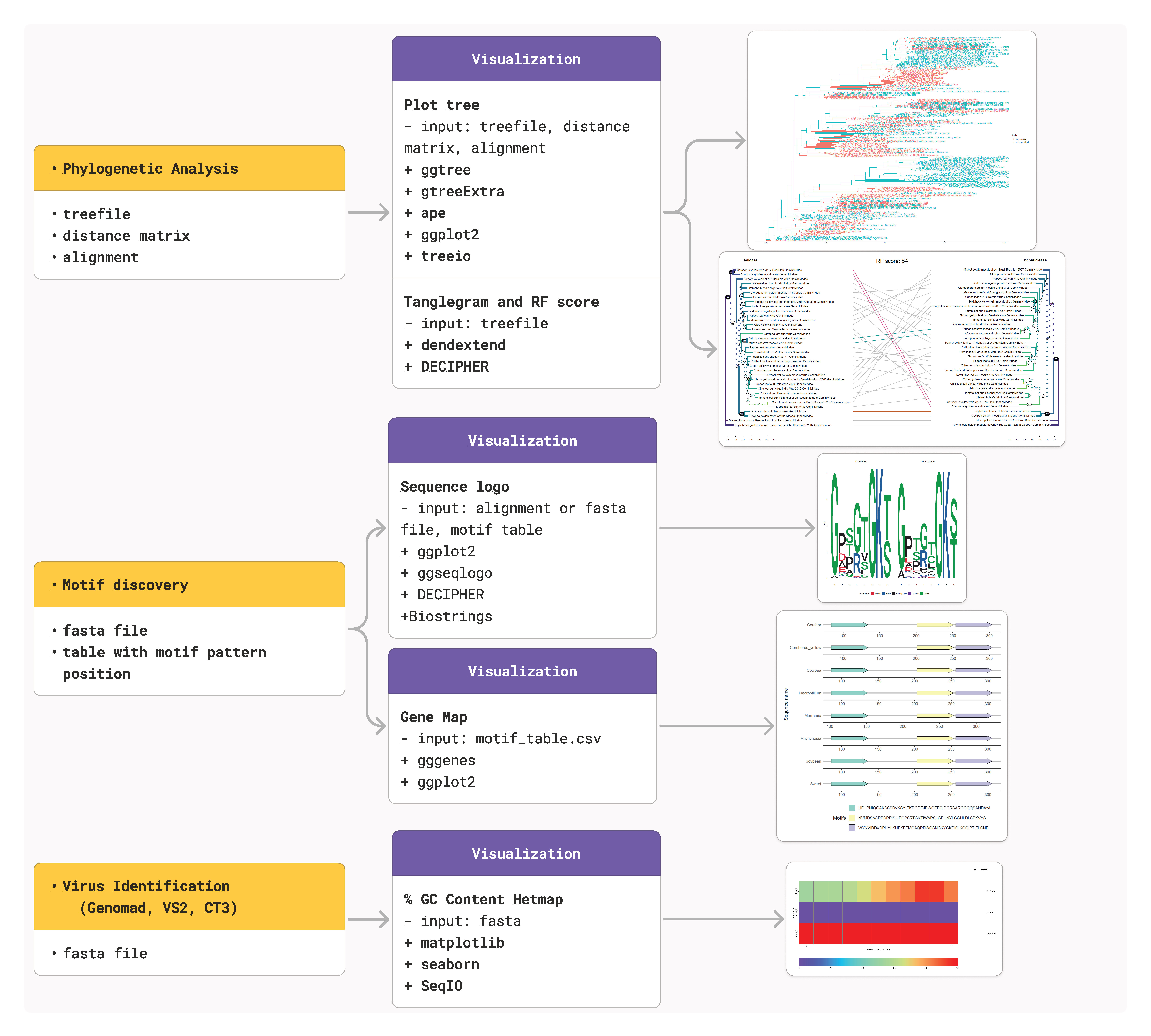
Overview
The visualization suite includes four primary modules:
Sequence logos for motif visualization
Phylogenetic trees with advanced customization options
Tanglegrams for comparative phylogenetic analysis
Motif mapping for genome-wide pattern distribution
Sequence Logo Generation
Purpose
Create information-rich sequence logos that display motif conservation patterns, nucleotide preferences, and positional information content.
Basic Usage
# From FASTA sequences
cressent seq_logo -i aligned_sequences.fasta \
-o visualization/ \
-n motif_logo.pdf \
--plot_title "CRESS Nonanucleotide"
# From motif table
cressent seq_logo -tb motif_positions.txt \
-o visualization/ \
-n discovered_motifs.pdf \
--width 12 \
--height 6
Key Features
Multi-row support for long sequences (automatic wrapping)
Grouped analysis by metadata categories
Information content or probability-based scaling
Publication-ready output with customizable dimensions
Advanced Customization
cressent seq_logo -tb motif_table.txt \
--plot_title "Viral Family Comparison" \
--split \
--metadata family_data.csv \
--group_label "virus_family" \
--ncol 3 \
--positions_per_row 40 \
--method "bits" \
--width 15 \
--height 10 \
-o grouped_logos/
Parameters
Parameter |
Description |
Default |
|---|---|---|
|
Logo title |
sequence_logo |
|
Dimensions in inches |
10/10 |
|
Scaling method (bits/prob) |
prob |
|
Positions before wrapping |
50 |
|
Auto-wrap threshold |
100 |
|
Group by metadata |
False |
|
Columns for grouped display |
Required with –split |
Output Examples
Single sequence logo:
Standard logo for conserved motifs
Information content visualization
Multi-row display for long sequences
Grouped logos:
Family-specific motif patterns
Comparative analysis across groups
Statistical significance indicators
Phylogenetic Tree Visualization
Purpose
Generate sophisticated phylogenetic tree visualizations with metadata integration, alignment display, and publication-quality formatting.
Basic Usage
# Simple tree plot
cressent plot_tree -t phylogeny.treefile \
-o tree_output/ \
--plot_name tree_analysis.pdf
# Tree with metadata
cressent plot_tree -t phylogeny.treefile \
--metadata_1 sequence_info.csv \
--metadata_2 name_mapping.tsv \
-o tree_output/ \
--color TRUE \
--tip_label "family"
Layout Options
Multiple tree layouts for different analytical needs:
# Circular tree
cressent plot_tree -t tree.treefile \
--layout circular \
--open_angle 90 \
--offset 0.2 \
-o circular_trees/
# Rectangular tree with alignment
cressent plot_tree -t tree.treefile \
--layout rectangular \
--alignment sequences.fasta \
--metadata_1 annotations.csv \
--color TRUE \
-o aligned_trees/
# Unrooted network
cressent plot_tree -t tree.treefile \
--layout unrooted \
--branch_length "branch.length" \
--fig_width 12 \
--fig_height 12 \
-o network_trees/
Distance Matrix Trees
Generate trees directly from distance matrices:
cressent plot_tree --dist_matrix sequences.mldist \
--metadata_1 annotations.csv \
--layout circular \
--color TRUE \
--tip_label "species" \
-o distance_trees/
Advanced Features
Feature |
Description |
Usage |
|---|---|---|
Metadata integration |
Color coding by groups |
|
Alignment display |
MSA alongside tree |
|
Custom labeling |
Flexible tip labels |
|
Branch scaling |
Different length metrics |
|
Size control |
Publication dimensions |
|
Parameters
Parameter |
Description |
Default |
|---|---|---|
|
Tree layout style |
rectangular |
|
Branch length method |
branch.length |
|
Circular tree opening |
0 |
|
Tip label offset |
0.14 |
|
Metadata column for tips |
family |
|
Color by groups |
TRUE |
|
Show tip labels |
TRUE |
Tanglegram Analysis
Purpose
Compare two phylogenetic trees through tanglegram visualization, highlighting topological differences and calculating Robinson-Foulds distances.
Basic Usage
cressent tanglegram --tree1 nucleotide_tree.treefile \
--tree2 protein_tree.treefile \
--label1 "Nucleotide Tree" \
--label2 "Protein Tree" \
-o tanglegram_output/ \
--name_tanglegram comparison.pdf
Features
Automatic tree comparison with Robinson-Foulds scoring
Edge highlighting for topological differences
Branch color coding for common subtrees
Dynamic sizing based on tree complexity
Publication formatting with customizable dimensions
Advanced Analysis
cressent tanglegram --tree1 rep_phylogeny.treefile \
--tree2 cap_phylogeny.treefile \
--label1 "Rep Protein Tree" \
--label2 "Capsid Protein Tree" \
--width 25 \
--height 15 \
--lab_cex 1.2 \
-o comparative_analysis/ \
--name_tanglegram rep_vs_cap.pdf
Parameters
Parameter |
Description |
Default |
|---|---|---|
|
Input tree files |
Required |
|
Tree labels |
Tree 1/Tree 2 |
|
Figure dimensions |
20/11 |
|
Label size |
1.5 |
|
Output filename |
tanglegram.pdf |
Output Features
RF score display quantifying tree distance
Common subtree highlighting in matching colors
Distinctive edge emphasis for conflicting relationships
Automatic layout optimization for clarity
Motif Mapping Visualization
Purpose
Create genome-wide visualizations showing motif distribution patterns, functional annotations, and structural relationships.
Input Compatibility
Supports multiple input formats with automatic detection:
# Prosite results
cressent motif_map_viz -f scanprosite_results.csv \
-o motif_maps/ \
--format prosite
# MEME motif table
cressent motif_map_viz -f motif_table.csv \
-o motif_maps/ \
--format motif_table
# Auto-detection
cressent motif_map_viz -f results.csv \
-o motif_maps/ \
--format auto
Visualization Types
Linear genome maps:
Sequence-by-sequence motif distribution
Position-accurate mapping
Color-coded motif types
Scalable for multiple sequences
Density plots:
Motif frequency analysis
Position distribution patterns
Comparative statistics
Multi-panel layouts
Heatmaps:
Presence/absence matrices
Quantitative motif counts
Hierarchical clustering options
Interactive color scaling
Detailed genome maps:
High-resolution motif positioning
Functional annotation integration
Publication-ready formatting
Comprehensive legends
Example Workflow
# Step 1: Generate motif data
cressent motif_discovery -i sequences.fasta \
-o motif_analysis/ \
--scanprosite
# Step 2: Create comprehensive visualizations
cressent motif_map_viz -f motif_analysis/motif_table.csv \
-o visualization/ \
--format auto
# Step 3: Generate Prosite visualizations
cressent motif_map_viz -f motif_analysis/scanprosite_results.csv \
-o visualization/ \
--format prosite
Output Gallery
Linear maps (genome_map_linear_*.png):
Horizontal sequence representations
Motif positions as colored rectangles
Coordinate systems with position labels
Multiple sequences in parallel tracks
Density analysis (motif_density_*.png):
Bar plots of motif counts per sequence
Histograms of positional distributions
Statistical summaries and trends
Comparative analysis panels
Heatmaps (motif_heatmap_*.png):
Matrix representations of motif presence
Color intensity indicating abundance
Clustering dendrograms (optional)
Annotation tracks for metadata
Detailed maps (detailed_genome_map_*.png):
High-resolution sequence tracks
Precise coordinate information
Functional annotation overlays
Professional publication formatting
Integrated Visualization Workflows
Complete Analysis Pipeline
# 1. Generate sequence data
cressent motif -i sequences.fasta \
-p "TAGTATTAC" \
--generate-logo \
--plot-title "Nonanucleotide Motif" \
-o complete_analysis/
# 2. Build phylogenetic trees
cressent build_tree -i aligned_sequences.fasta \
-o complete_analysis/ \
-B 1000
cressent plot_tree -t complete_analysis/*.treefile \
--metadata_1 sequence_metadata.csv \
--layout circular \
--color TRUE \
-o complete_analysis/
# 3. Comparative phylogenetics
cressent tanglegram --tree1 rep_tree.treefile \
--tree2 cap_tree.treefile \
--label1 "Rep Proteins" \
--label2 "Capsid Proteins" \
-o complete_analysis/
# 4. Motif distribution analysis
cressent motif_map_viz -f complete_analysis/motif_table.csv \
-o complete_analysis/
Publication-Ready Output
# High-quality figures for publication
cressent seq_logo -tb motif_positions.txt \
--plot_title "Conserved Nonanucleotide" \
--method "bits" \
--width 8 \
--height 4 \
-n Figure_1A.pdf \
-o publication_figures/
cressent plot_tree -t phylogeny.treefile \
--metadata_1 annotations.csv \
--layout rectangular \
--alignment sequences.fasta \
--fig_width 10 \
--fig_height 8 \
--plot_name Figure_2.pdf \
-o publication_figures/
cressent tanglegram --tree1 tree1.treefile \
--tree2 tree2.treefile \
--width 12 \
--height 8 \
--name_tanglegram Figure_3.pdf \
-o publication_figures/
Best Practices
Design Principles
Consistency in color schemes and styling
Clarity in labeling and legends
Scalability for different output formats
Accessibility with colorblind-friendly palettes
Technical Considerations
Resolution appropriate for intended use
File formats suitable for target applications
Size optimization for web or print
Reproducibility through parameter documentation
Quality Control
Preview outputs before final generation
Validate data integrity in visualizations
Test different parameter combinations
Document successful parameter sets
Troubleshooting
Common Issues
R package dependencies:
Ensure all required packages are installed
Check version compatibility
Update packages if necessary
Large dataset visualization:
Reduce data complexity where possible
Increase memory allocation
Use sampling for preview purposes
Color scheme problems:
Test with colorblind simulation tools
Use established palettes (RColorBrewer)
Maintain sufficient contrast
Performance Optimization
Memory management:
Process large datasets in chunks
Clear R workspace between analyses
Monitor system resources
Rendering speed:
Use appropriate resolution settings
Optimize data structures
Consider vector vs. raster formats
Output Validation
Quality checks:
Verify data accuracy in plots
Check scaling and proportions
Validate legends and labels
Test across different viewers/platforms