Sequence Alignment Module
The align module performs multiple sequence alignment using MAFFT and trimming using TrimAl. It can work with both standalone sequences and database-integrated alignments for phylogenetic analysis.
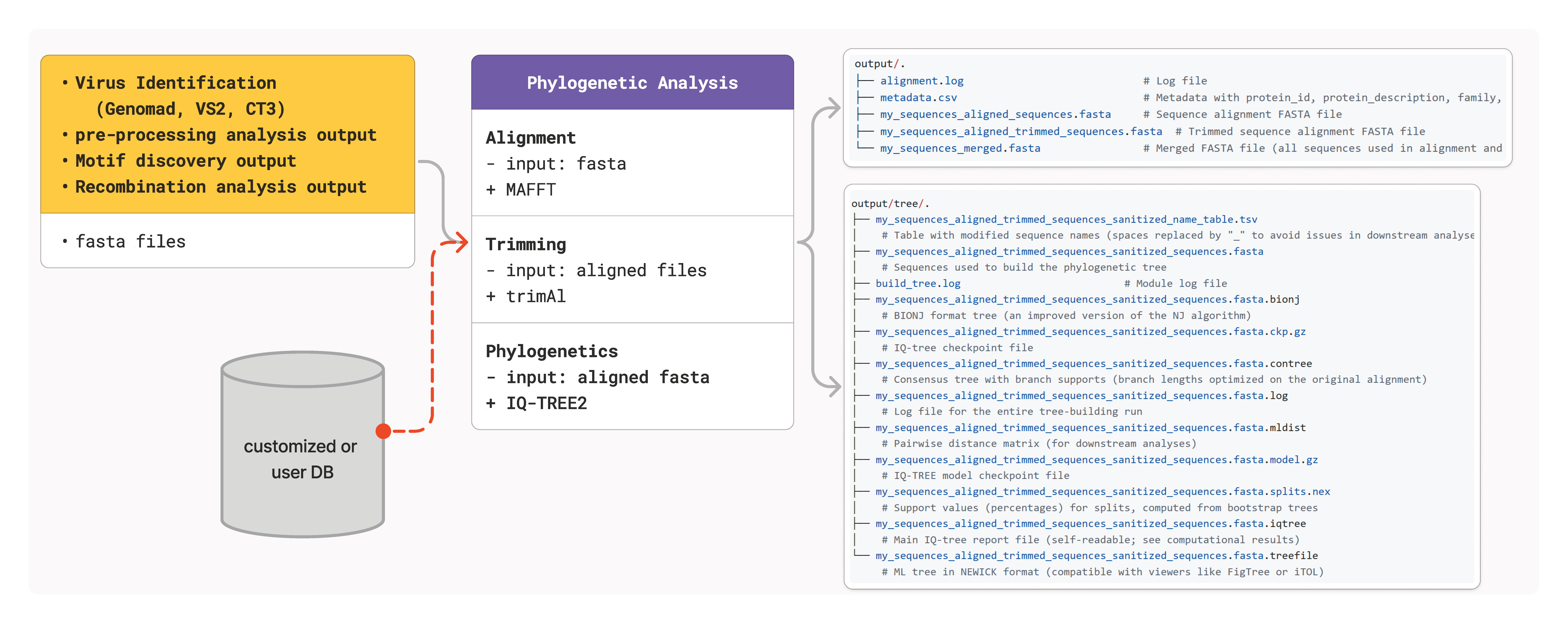
Overview
The alignment module is essential for downstream phylogenetic analysis and serves as the foundation for:
Phylogenetic tree construction
Recombination detection
Motif analysis
Comparative genomics studies
Workflow
The align module follows a structured workflow:
Input Validation: Validates FASTA format and sequence integrity
Database Integration (optional): Merges input sequences with reference database sequences
Metadata Generation: Creates comprehensive metadata for all sequences
Multiple Sequence Alignment: Uses MAFFT with optimized parameters for protein/nucleotide sequences
Alignment Trimming: Removes poorly aligned regions using TrimAl
Usage
Basic Alignment
Align sequences without database integration:
cressent align \
--threads 24 \
--input_fasta sequences.fasta \
-o output/alignment
Database-Integrated Alignment
Align sequences with viral family database for enhanced phylogenetic context:
cressent align \
--threads 24 \
--input_fasta sequences_reps.faa \
--db_family "Naryaviridae" \
--protein_type reps \
--db_path databases/ \
-o output/alignment_with_db
Custom Database Alignment
Use a your custom database for alignment:
cressent align \
--threads 24 \
--input_fasta sequences.fasta \
--db_family "custom" \
--custom_aa custom_sequences.faa \
-o output/custom_alignment
How do I build custom reference databases?
Use the db_buildermodule:
The taxonomic list is here.
cressent db_builder \
-t taxonomy_file.csv \
-l Genus \
-s "YourVirusGenus" \
-o custom_database \
-e your.email@example.com
The final database would contain:
custom_database/YourVirusGenus/
├── annotated/
│ ├── caps/ # Capsid proteins by cluster (if there sequences were found)
│ └── reps/ # Replication proteins by cluster (if there sequences were found)
├── unannotated/ # Unclassified ORFs (if there sequences were found)
├── cd_hit/ # CD-HIT output
├── db_builder.log # log file
├── diamond/ # diamond output
├── mcl/ # MCL output
├── raw_aa # Family-specific raw protein sequences
Ensure your taxonomy file contains proper ICTV classifications and accession numbers.
Parameters
Required Parameters
-i, --input_fasta: Input FASTA file containing sequences to align-o, --output: Output directory for alignment results
Optional Parameters
-t, --threads: Number of CPU threads (default: 1)--mafft_ep: MAFFT alignment accuracy parameter (default: 0.123)--gap_threshold: TrimAl gap threshold for trimming (default: 0.2)
Database Parameters
--db_family: Viral family name(s) for database selection or ‘all’ for complete database--db_path: Path to the database directory--protein_type: Specify ‘reps’ or ‘caps’ for protein-specific databases--custom_aa: Path to custom amino acid database file
Output Files
The align module generates several important output files:
Primary Outputs
<prefix>_aligned_sequences.fasta: Raw MAFFT alignment<prefix>_aligned_trimmed_sequences.fasta: Trimmed alignment ready for phylogenetic analysismetadata.csv: Comprehensive sequence metadata including family assignments
Metadata Structure
The metadata file contains the following columns:
Column |
Description |
|---|---|
protein_id |
Unique sequence identifier |
protein_description |
Full sequence description |
family |
Assigned viral family |
scientific_name |
Source organism name |
protein_name |
Protein function/name |
source |
Origin (input or database) |
Best Practices
Sequence Preparation
Ensure sequence quality: Remove sequences with excessive ambiguous nucleotides
Check sequence orientation: All sequences should be in the same orientation
Validate functional domains: For proteins, ensure sequences contain expected functional domains
Parameter Optimization
Thread usage: Use available CPU cores but monitor memory usage
Gap threshold: Lower values (0.1-0.3) for conserved sequences, higher (0.4-0.6) for divergent sequences
Database selection: Use family-specific databases when available for better phylogenetic signal
Quality Control
After alignment, check:
Alignment length: Should retain sufficient positions for phylogenetic analysis
Sequence coverage: Most sequences should span the majority of the alignment
Conserved regions: Key functional domains should be well-aligned
Integration with Other Modules
The align module outputs are directly compatible with:
build_tree: For phylogenetic tree construction
recombination: For recombination detection analysis
motif: For motif discovery and analysis
plot_tree: For tree visualization with alignment context
Troubleshooting
Common Issues
Memory Errors Use fewer threads or reduce dataset size. Consider clustering sequences first.
Poor Alignment Quality
Adjust --mafft_ep parameter or check input sequence quality.
Database Integration Failures Verify database path and family names. Ensure database files exist.
Empty Output Check input file format and sequence validity. Review log files for specific errors.
Performance Tips
Large datasets: Use sequence clustering before alignment
Memory optimization: Reduce thread count if memory is limited
Speed optimization: Use family-specific databases instead of ‘all’
Example Workflow
Here’s a complete example for capsid protein alignment:
# Basic alignment for tree building
cressent align \
--threads 24 \
--input_fasta capsid_proteins.faa \
-o analysis/caps_align
# Database-integrated alignment for comprehensive phylogeny
cressent align \
--threads 24 \
--input_fasta capsid_proteins.faa \
--db_family "Circoviridae" "Genomoviridae" \
--protein_type caps \
--db_path /path/to/databases \
-o analysis/caps_align_with_db
# Build tree from alignment
cressent build_tree \
-i analysis/caps_align_with_db/capsid_proteins_aligned_trimmed_sequences.fasta \
-o analysis/caps_tree