Phylogenetic Analysis
Phylogenetic analysis in CRESSENT combines high-quality multiple sequence alignment with robust tree construction methods to reconstruct evolutionary relationships among ssDNA viruses. This integrated approach ensures reliable phylogenetic inferences for comparative studies.
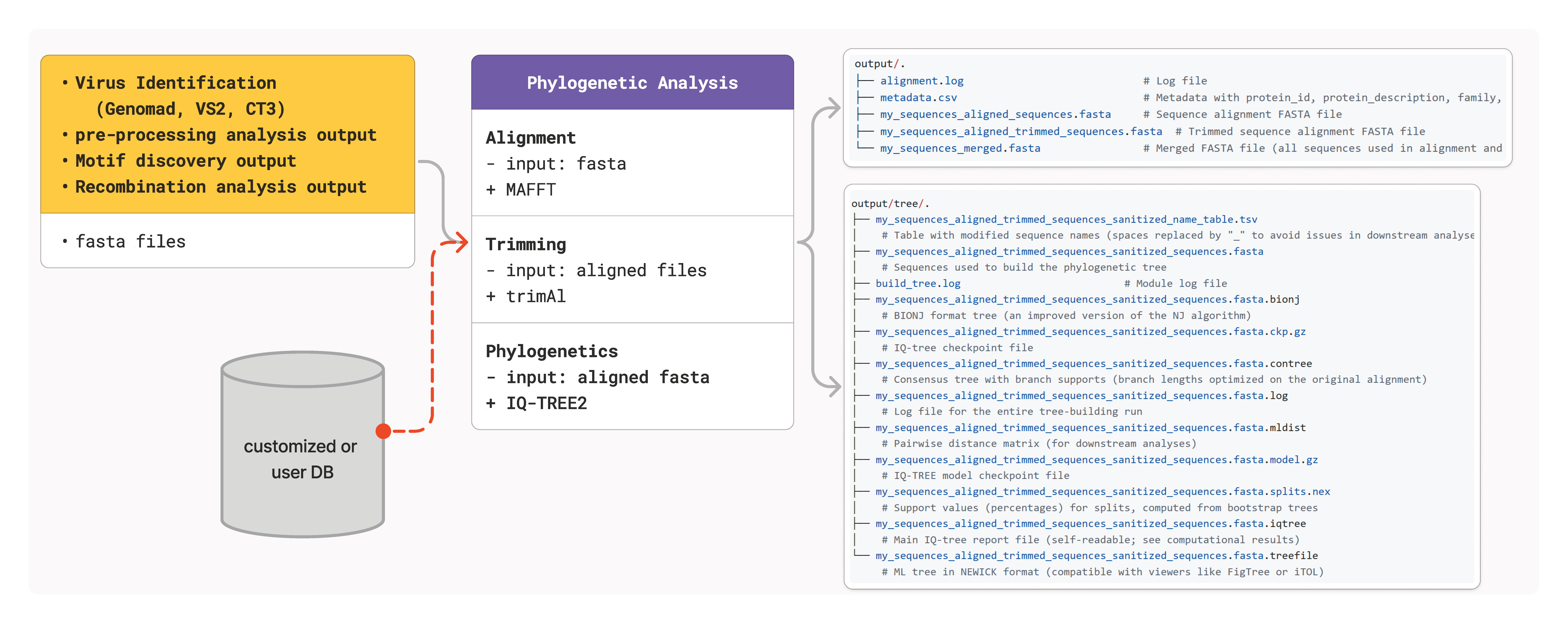
Overview
CRESSENT’s phylogenetic analysis pipeline consists of two main components:
Sequence Alignment: Multiple sequence alignment using MAFFT with database integration
Tree Construction: Maximum likelihood phylogenetic inference using IQ-TREE
This integrated workflow provides publication-ready phylogenetic trees with comprehensive statistical support.
Workflow Components
Sequence Alignment Module
The alignment module performs several critical functions:
Input Processing
FASTA format validation and sequence quality checks
Automatic sequence type detection (nucleotide vs. protein)
Sequence name sanitization for downstream compatibility
Database Integration
Integration with viral family-specific reference databases
Metadata generation for enhanced phylogenetic context
Custom database support for specialized analyses
Alignment Generation
MAFFT-based multiple sequence alignment with optimized parameters
TrimAl-based trimming to remove poorly aligned regions
Quality assessment and validation
Tree Construction Module
The tree building module provides robust phylogenetic inference:
Model Selection
Automatic evolutionary model selection using ModelFinder
Support for user-specified models for targeted analyses
Model adequacy testing and validation
Tree Inference
Maximum likelihood tree construction using IQ-TREE
Bootstrap analysis for statistical support assessment
Branch length optimization and topology testing
Output Generation
Newick format trees compatible with visualization tools
Comprehensive log files with analysis statistics
Name mapping tables for result interpretation
Basic Phylogenetic Workflow
Step 1: Sequence Alignment
# Basic protein alignment
cressent align \
--threads 24 \
--input_fasta rep_proteins.faa \
-o analysis/alignment
# Database-integrated alignment for phylogenetic context
cressent align \
--threads 24 \
--input_fasta rep_proteins.faa \
--db_family "Naryaviridae" \
--protein_type reps \
--db_path databases/ \
-o analysis/alignment_with_db
Database can be downloaded from ![]()
Step 2: Tree Construction
# Automatic model selection and tree building
cressent build_tree \
-i analysis/alignment/rep_proteins_aligned_trimmed_sequences.fasta \
-o analysis/tree \
-m MFP \
--bootstrap 1000
# Fast tree with specified model
cressent build_tree \
-i analysis/alignment/rep_proteins_aligned_trimmed_sequences.fasta \
-o analysis/tree \
-m WAG+G4 \
--bootstrap 100
💡💡 Evolutionary models have already been precomputed by ModelFinder from IQ-TREE2, which can reduce both processing time and computational cost in this module. 💡💡
Advanced Phylogenetic Analyses
Family-Level Comparative Analysis
⚠️⚠️ CAUTION !!! ⚠️⚠️
Since CRESSENT was developed and tailored specifically for family-level ssDNA virus analysis, expanding the database to include multiple additional families may substantially increase processing time and computational cost. Therefore, use the full database with caution.
We recommend performing analyses at the family level whenever possible.
# 1. Align with complete family database
cressent align \
--threads 32 \
--input_fasta novel_sequences.faa \
--db_family "Circoviridae" "Genomoviridae" "Smacoviridae" \
--protein_type reps \
--db_path viral_databases/ \
-o analysis/family_comparison
# 2. Build comprehensive phylogeny
cressent build_tree \
-i analysis/family_comparison/novel_sequences_aligned_trimmed_sequences.fasta \
-o analysis/family_tree \
-m MFP \
--bootstrap 1000 \
--extra_args '-bb 1000 -alrt 1000'
Domain-Specific Phylogenetics
For protein domain analysis:
# 1. Split sequences at conserved motif
cressent motif \
-i full_proteins_aligned.fasta \
-o domain_analysis \
-p ".{5}GK[TS].{4}" \
--split-sequences
# 2. Analyze N-terminal domain
cressent align \
--threads 24 \
--input_fasta domain_analysis/split_sequences_1.fasta \
-o analysis/nterminal_domain
cressent build_tree \
-i analysis/nterminal_domain/split_sequences_1_aligned_trimmed_sequences.fasta \
-o analysis/nterminal_tree
# 3. Analyze C-terminal domain
cressent align \
--threads 24 \
--input_fasta domain_analysis/split_sequences_2.fasta \
-o analysis/cterminal_domain
cressent build_tree \
-i analysis/cterminal_domain/split_sequences_2_aligned_trimmed_sequences.fasta \
-o analysis/cterminal_tree
Nucleotide vs. Protein Phylogenies
Compare phylogenetic signal at different levels:
# Nucleotide-based phylogeny
cressent align \
--threads 24 \
--input_fasta coding_sequences.fna \
-o analysis/nucleotide_align
cressent build_tree \
-i analysis/nucleotide_align/coding_sequences_aligned_trimmed_sequences.fasta \
-o analysis/nucleotide_tree \
-m GTR+G4
# Protein-based phylogeny
cressent align \
--threads 24 \
--input_fasta protein_sequences.faa \
-o analysis/protein_align
cressent build_tree \
-i analysis/protein_align/protein_sequences_aligned_trimmed_sequences.fasta \
-o analysis/protein_tree \
-m WAG+G4
Quality Assessment
Alignment Quality Metrics
Evaluate alignment quality before tree construction:
Coverage Assessment
Sequence coverage across alignment length
Gap distribution analysis
Conserved region identification
Compositional Analysis
Amino acid/nucleotide composition
Substitution saturation assessment
Phylogenetic informativeness
Tree Support Evaluation
Assess phylogenetic reliability:
Bootstrap Support
Values ≥70% indicate reliable relationships
Values ≥95% represent very strong support
Focus interpretation on well-supported clades
Branch Length Analysis
Reasonable evolutionary distances
Detection of unusually long branches
Clock-like behavior assessment
Parameter Optimization
Alignment Parameters
For Highly Similar Sequences (>90% identity):
--mafft_ep 0.1 --gap_threshold 0.1
For Moderately Divergent Sequences (70-90% identity):
--mafft_ep 0.123 --gap_threshold 0.2 # Default values
For Highly Divergent Sequences (<70% identity):
--mafft_ep 0.5 --gap_threshold 0.4
Tree Construction Parameters
For Quick Analysis:
-m WAG+G4 --bootstrap 100
For Publication-Quality Analysis:
-m MFP --bootstrap 1000 --extra_args '-bb 1000 -alrt 1000'
For Large Datasets (>500 sequences):
-m GTR+G4 --bootstrap 100 --extra_args '-fast'
Integration with Other Modules
Recombination-Aware Phylogenetics
# 1. Detect recombination first
cressent recombination \
-i aligned_sequences.fasta \
-o recombination_analysis \
--all
# 2. Remove recombinant sequences if needed
# 3. Build phylogeny with cleaned dataset
cressent build_tree \
-i cleaned_alignment.fasta \
-o clean_phylogeny
Visualization Integration
# Create publication-ready tree figures
cressent plot_tree \
--tree analysis/tree/sequences_aligned_trimmed_sequences.treefile \
-o analysis/visualization \
--metadata_1 analysis/alignment/metadata.csv \
--layout circular \
--fig_width 12 --fig_height 10
Common Applications
Taxonomic Classification
Place unknown sequences in phylogenetic context:
# Include reference sequences from known taxa
cressent align \
--input_fasta unknown_viruses.faa \
--db_family "all" \
--protein_type reps \
-o taxonomic_placement
cressent build_tree \
-i taxonomic_placement/unknown_viruses_aligned_trimmed_sequences.fasta \
-o taxonomic_tree \
-m MFP
Host-Virus Coevolution
Analyze parallel phylogenies:
# Build virus phylogeny
cressent build_tree \
-i virus_alignment.fasta \
-o virus_tree
# Compare with host phylogeny using tanglegram
cressent tanglegram \
--tree1 virus_tree/virus_alignment.treefile \
--tree2 host_phylogeny.tre \
--label1 "Virus" \
--label2 "Host" \
-o coevolution_analysis
Outbreak Investigation
Track viral transmission:
# High-resolution phylogeny for outbreak strains
cressent align \
--input_fasta outbreak_strains.fna \
-o outbreak_analysis
cressent build_tree \
-i outbreak_analysis/outbreak_strains_aligned_trimmed_sequences.fasta \
-o outbreak_tree \
-m GTR+G4 \
--bootstrap 1000
Best Practices
Input Preparation
Sequence Quality: Remove sequences with excessive ambiguous characters
Homology Assessment: Ensure sequences represent homologous regions
Length Filtering: Remove sequences that are too short or too long
Functional Validation: Verify sequences contain expected functional domains
Parameter Selection
Model Choice: Use MFP for model selection, specific models for speed
Bootstrap Replicates: 100 for preliminary analysis, 1000 for publication
Thread Usage: Balance CPU cores with available memory
Database Selection: Use family-specific databases when available
Result Interpretation
Support Values: Focus on relationships with ≥70% bootstrap support
Branch Lengths: Examine for biological reasonableness
Topology: Validate against known biological relationships
Outgroup: Include appropriate outgroup sequences when possible
Troubleshooting
Common Issues
Poor Alignment Quality
Check input sequence homology
Adjust MAFFT parameters for sequence divergence
Consider protein vs. nucleotide alignment
Tree Construction Failures
Verify alignment has sufficient informative sites
Check for identical sequences
Try simpler evolutionary models
Low Bootstrap Support
Increase bootstrap replicates
Check for conflicting phylogenetic signal
Consider recombination detection
Performance Optimization
Memory Management
Monitor RAM usage during large analyses
Reduce thread count if memory-limited
Use clustering to reduce dataset size
Speed Optimization
Use specific models instead of model selection
Reduce bootstrap replicates for preliminary analysis
Employ fast approximation methods for large datasets
Example Complete Workflow
#!/bin/bash
# Complete phylogenetic analysis workflow
echo "Starting phylogenetic analysis..."
# Set up directories
mkdir -p phylogeny/{alignment,tree,visualization}
# 1. High-quality alignment with database context
cressent align \
--threads 32 \
--input_fasta input_sequences.faa \
--db_family "target_family" \
--protein_type reps \
--db_path databases/ \
-o phylogeny/alignment
# 2. Robust tree construction
cressent build_tree \
-i phylogeny/alignment/input_sequences_aligned_trimmed_sequences.fasta \
-o phylogeny/tree \
-m MFP \
--bootstrap 1000
# 3. Create publication figures
cressent plot_tree \
--tree phylogeny/tree/input_sequences_aligned_trimmed_sequences.treefile \
-o phylogeny/visualization \
--metadata_1 phylogeny/alignment/metadata.csv \
--layout circular \
--fig_width 15 --fig_height 12 \
--plot_name family_phylogeny.pdf
echo "Phylogenetic analysis complete!"
echo "Tree: phylogeny/tree/*.treefile"
echo "Visualization: phylogeny/visualization/*.pdf"
This comprehensive phylogenetic analysis framework provides the foundation for robust evolutionary studies of ssDNA viruses, from basic tree construction to sophisticated comparative genomics.